Epilepsia refratárias da infância
A epilepsia é muito comum na infância, com uma prevalência de aproximadamente 5/1000 com maior frequência na faixa etária de 0 a 9 anos de idade. Grande parte das epilepsias de início na infância tem um prognóstico favorável.(20,21) O tratamento adequado com uma droga antiepiléptica tem êxito em aproximadamente 50% dos pacientes, a adição de uma droga antiepiléptica possibilitará somar mais 25% de pacientes controlados, portanto chegando a índices de 75%.(38,48,49) No entanto a exata prevalência de epilepsia de difícil controle não é bem conhecida.
Aproximadamente 75% de todos pacientes com um diagnóstico de epilepsia entram em remissão (ficam livres de crises por 5 ou mais anos) e mais do que 50% entram em remissão no primeiro ano que segue ao diagnóstico (21,30,38). Estima-se que cerca de 20 a 30% dos casos pode ser de difícil controle medicamentoso. (20,22,68)
Indicadores de refratariedade
Poucas informações estão disponíveis sobre os indicadores de epilepsia de difícil controle na criança. Os fatores de resistência ao tratamento medicamentoso nas epilepsias da infância estão relacionados ao início precoce antes dos 2 anos de vida, a presença de estado de mal epiléptico prévio e a presença alteração estrutural cerebral como os distúrbios de migração neuronal e as malformações vasculares.(9,48) Outros fatores associados também são referidos como indicativos da refratariedade como a presença de deterioração neuropsicológica e anormalidades no exame neurológico inicial, crises epilépticas do tipo tônica, atônica, espasmos infantis, presença de vários tipos de crises epilépticas inaugurando o quadro, elevada freqüência de crises no início da epilepsia, história clínica de longa duração, falha de tratamentos prévios e EEG inicial francamente anormal com atividade de base alentecida e paroxismos multifocais.(10,65,69) É importante ressaltar que a refratariedade pode também dever-se a falha no diagnóstico diferencial com as crises pseudo-epilépticas como as que acompanham perda de fôlego, síncopes vaso-vagais, síndrome do QT longo, distúrbios do sono e dos movimentos involuntários. Crises frequentes e resistentes ao tratamento medicamentosos podem fazer parte do contexto sintomatológico de encefalopatias progressivas como a panencefalite subaguda esclerosante, a forma infantil da doença de Huntington, a doença de Lafora , a lipofuccinose ceróide e algumas formas precoces das encefalopatias mitocondriais.(38,48) Ainda não está bem estabelecido se a utilização de um tratamento efetivo e precoce pode levar a uma remissão espontânea das crises epilépticas ou mesmo da própria epilepsia. Em países em desenvolvimento, distúrbios perinatais e infecções do sistema nervoso central são muito comuns e também são responsáveis por uma alta porcentagem de epilepsia parcial sintomática levando a uma maior prevalência de epilepsia de difícil controle na infância.(21,30,38,49)
Conceito de Epilepsia refratária
Existe uma terminologia muito variável para o grupo de epilepsia da infância com evolução desfavorável como: epilepsias graves, epilepsias refratárias, epilepsias de difícil tratamento, epilepsias severas, epilepsias de difícil controle, epilepsias intratáveis, epilepsias catastróficas.(10,65,69) Nenhum destes conceitos repousam sobre um consenso clínico ou etimológico. Assim, Berg et al. (1996) referem que uma criança é considerada portadora de epilepsia de difícil controle medicamentoso quando apresenta pelo menos uma crise epiléptica por mês por um período mínimo de 2 anos e que durante esse período três diferentes drogas antiepilépticas foram utilizadas em monoterapia ou politerapia. (9)
O conceito de epilepsia de difícil controle na infância engloba também o critério de aceitação individual de crises epilépticas e a repercussão das crises sobre a qualidade de vida da criança, incluindo neste contexto as repercussões neuropsicológicas, comportamentais e os efeitos disruptivos das crises na dinâmica familiar e social da criança.
As síndromes epilépticas refratárias da infância constituem-se em um grupo bastante polimorfo tanto em suas manifestações clínicas como na variabilidadede da expressão individual. Fatores relacionados com a maturação cerebral e neuromodulação, bem como com os efeitos das descargas paroxísticas sobre determinados circuitos neuronais em desenvolvimento são responsáveis pelas diferentes expressões das epilepsias na infância quando comparados a população epiléptica adulta.(49,54) Tais fatores apontam para a necessidade do diagnóstico precoce na delimitação de síndromes epilépticas relacionadas a um pior prognóstico. Assim o conceito das encefalopatias epilépticas centra-se no fato de que as próprias crises epilépticas ou mesmo as descargas interictais podem ser determinantes no desenvolvimento dos déficit cognitivos e motores associados às epilepsias graves , o que se esboça conceitualmente como “plasticidade cerebral negativa”.(2,28,51) Neste sentido prevenir e tratar as crises de início precoce pode minimizar o impacto neuropsicológico e as sequelas associadas a epilepsias de difícil controle. (65,67,69)
O grande número de crises epilépticas de difícil controle medicamentoso relaciona-se a fatores neuromaturacionais e fisiológicos próprios do cérebro em desenvolvimento como a elevada excitabilidade, a reorganização de conexões excitatórias recorrentes em áreas susceptíveis a hipoxia e isquemia como o hipocampo e mesmo à diminuição de circuitos inibitórios mediados pelo GABA ou ainda, a diferenças no desenvolvimento de circuitos inibitórios em áreas subcorticais.
Contexto Clínico
A resposta ao tratamento com drogas antiepilépticas e o prognóstico dos diversos tipos de crises epilépticas, diferem de acordo com a síndrome epiléptica envolvida Assim o delineamento de síndromes epilépticas específicas permite dar precisão ao diagnóstico e ao prognóstico da epilepsia. Na infância e adolescência há um grupo de síndromes com um bom prognóstico como: as convulsões neonatais benignas, epilepsia mioclônica benigna na infância, epilepsia de ausência típica, epilepsia com crises generalizadas tônico-clônicas, epilepsia parcial rolândica, epilepsia parcial benigna com paroxismos occipitais, epilepsia mioclônica juvenil e epilepsia do despertar.(18,19,30) As etiologias associadas a epilepsia de difícil controle na infância são múltiplas esclerose mesial temporal, leucomalácia, displasia cortical, hamartomas, tumores gliais, facomatoses, anóxia perinatal, meningencefalite, traumatismo crânio-encefálico, malformação arterio-venosa, microcefalia e encefalite crônica, mas em muitos casos a etiologia não é determinada mesmo após investigação exaustiva.(9,12).
Não só o contexto sindrômico mas particularmente alguns tipos de crises epilépticas têm prognóstico reservado. Neste grupo destacam-se os espasmos infantis, as crises de ausência atípica, as crises tônicas, as crises atônico-mioclônicas de “drop atacks” e as crises parciais complexas, ressaltando-se, que na infância as crises parciais complexas tem prognóstico relativamente mais favorável do que o observado em adultos.(26,30) O grupo de difícil controle medicamentoso, embora heterogêneo em relação a combinação de diferentes crises epilépticas e etiologias, é em quase a totalidade dos casos constituído por crianças que, além de sofrer os efeitos adversos e somatórios das várias DAE em regime de politerapia, são cercados de graves problemas de adaptação psico-social diante de uma doença crônica, refratária e debilitante.(54) A esse grupo de difícil controle medicamentoso daremos especial atenção em nosso trabalho. Ressaltamos que em muitas ocasiões o contexto clínico e sintomatológico não nos possibilita a delimitação de síndromes epilépticas específicas. (18,19,30).
Iremos a seguir abordar os principais quadros sindrômicos associado a epilepsia de difícil controle medicamentoso por ordem de idade de aparecimento .
Encefalopatia epiléptica com padrão de “surto-supressão”
A encefalopatia com padrão de “surto-supressão” está associada a duas entidades clínicas distintas. A encefalopatia infantil precoce inicialmente descrita por Ohtahara e a encefalopatia mioclônica precoce descrita por Dalla-Bernardina e Aicardi. (1,26,27)
A encefalopatia epiléptica infantil conhecida como síndrome de Ohtahara é uma entidade rara com prevalência de 0,2% das epilepsias na infância.(49) Manifesta-se precocemente, sempre dentro dos três meses de vida, e habitualmente nos primeiros 20 dias de vida. As crises epilépticas predominantes são os espasmos tônicos, de difícil controle medicamentoso, podendo estar presentes crises focais multi-segmentares. Na síndrome de Ohtahara não ocorrem crises mioclônicas. As etiologias são variadas, estando principalmente relacionadas a malformações do Sistema Nervoso Central como as disgenesias que acompanham a síndrome de Aicardi.(51) Distúrbios metabólicos como a deficiência de citocromo-oxidase são também relatados.(13,74) Observa-se quadro de importante atraso do desenvolvimento neuropsicomotor e o EEG mostra um padrão surto-supressão que não se altera com o estado de vigília da criança.(1,17) Fig. 1.
Fig.1 Eletrencefalograma característico de padrão surto-supressão na S. Otahara
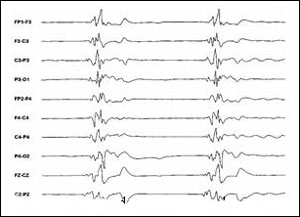
Metade dos pacientes com síndrome de Ohtahara evoluem para o óbito num período de 10 anos e 25% dos pacientes morrem nos dois primeiros anos de vida em consequências clínicas de crises intratáveis. Os que sobrevivem com freqüência evoluem com sequelas graves como retardo mental, espasticidade e paresias. Alguns casos desenvolvem a síndrome de West na idade de 4 a 6 meses e posteriormente a síndrome de Lennox-Gastaut. As crises epilépticas são refratárias tanto às drogas antiepilépticas habituais como também às novas drogas antiepilépticas. O ACTH tem um efeito limitado sobre as crises epilépticas tônicas e parciais.
Na encefalopatia mioclônica precoce as crises epilépticas iniciam-se no período neonatal e caracterizam-se por mioclonias parciais erráticas e fragmentadas, mioclonias maciças, crises motoras parciais e espasmos tônicos geralmente a partir do 3 ou 4 mês de vida. O EEG mostra surto-supressão com paroxismos de ondas agudas e atividade lenta seguidos de atenuação de 3 a 10 segundos. Tal padrão tende a ser totalmente substituído por hipsarritmia ou padrão multifocal com a idade de 3 a 5 meses.(17,51,74) A apresentação clínica é variável sendo que algumas crianças podem apresentar-se sem anormalidades ao nascimento, com rápida deterioração ou apresentam encefalopatia franca desde o nascimento. As crises epilépticas são de difícil controle medicamentoso e frequentemente ocorre involução neuropsicomotora. A grande maioria das crianças morrem antes de um ano de vida. As etiologias são variadas podendo estar relacionadas a anóxia neonatal, a malformações cerebrais congênitas e erros inatos do metabolismo como a hiperglicinemia não-cetótica. A presença de mioclonias na encefalopatia mioclônica precoce diferencia esta entidade da síndrome de Ohtahara.(51,74)
Síndrome de West
A síndrome de West consiste de uma tríade clínica e eletrencefalográfica caracterizada por espasmos infantis, atraso ou deterioração do desenvolvimento neuropsicomotor e padrão eletrencefalográfico característico conhecido como hipsarritmia, A idade de início ocorre entre 3 a 7 meses, com pico na idade de 5 meses. Os espasmos infantis ocorrem na frequência de 1 para 3.000 a 4.000 crianças e é encontrada em 9% de todas crianças com epilepsia e apresentam discreto predomínio no sexo masculino (5,22,49)
Os espasmos infantis constituem-se de movimentos rápidos, geralmente em salva acometendo a musculatura cervical, facial e dos membros superiores e inferiores com apresentação polimorfa de acordo com a musculatura envolvida. Assim podem ser em flexão, extensão, misto com abertura ou flexão dos membros superiores, tendo apresentação simétrica ou assimétrica, unilateral ou bilateral.. Espasmos assimétricos geralmente associam-se a anormalidades orgânicas estruturais como displasias ou malformações cerebrais.(31) Apresentam-se caracteristicamente como movimentos rápidos de 1 a 15 segundos de duração, presentes tanto em vigília como no sono, mais comumente no início do sono e despertar, podendo associar-se a fenômenos autonômicos como taquicardia, cianose, apnéia, automatismos de choro, riso ou grito. Os espasmos podem também ser .acompanhados de como movimentos sutis restritos a musculatura facial ou ocular com nistagmo ou flutter ocular. A brevidade dos episódios, a apresentação em salvas e a semelhança dos episódios a cólicas do lactente ou reflexo de Moro, muitas vezes retardam o diagnóstico precoce e contribuem para deterioração psicomotora, presente em praticamente 95 % dos casos não tratados.(5,21). A hipsarritmia (Fig 2) é caracterizada por uma mistura caótica de ondas lentas de alta amplitude, na freqüência de 1 a 7 c/s, com ondas agudas (sharp) e pontas (spikes) que variam em amplitude, morfologia, duração e localização. O padrão hipsarritmico pode ser interpretado como a expressão de 3 características de acordo com a idade-dependente do cérebro imaturo: a excitabilidade, alta tendência a difusão dos paroxismos e a imaturidade de sincronização das espículas nos dois hemisférios cerebrais.
Fig 2. Hipsarritmia (vigília)
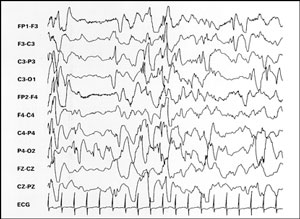
Estudos animais mostram que a hiperexcitabilidade transitória do córtex imaturo ocorre devido a representação exacerbada de receptores NMDA (excitatórios) e às fibras axonais colaterais que estão transitoriamente aumentadas. A imaturidade cerebral reflete-se também na incompleta mielinização que determina uma condução interhemisférica mais lenta resultando muitas vezes em descargas assíncronas em ambos hemisférios cerebrais.. Após 1 ano de vida a progressiva mielinização leva a um aumento da condução e os paroxismos tornam-se mais sincronizados. A hipsarritmia pode resultar na difusão de anormalidades focais sendo consequente a uma generalização secundária ou quando não existe lesão estrutural a hiperexcitabilidade resultaria de um fenômeno generalizado primário. (17)
Assim o EEG pode expressar anormalidades localizadas, lentificação focal e hipssaritmia assimétrica. Estudos com videotelemetria e monitorização eletrencefalográfica contínua tem valorizado as manifestações clínicas assimétricas como desvio cefálico e ocular lateralizado e as alterações eletrencefalográficas interictais como paroxismos focais, alentecimento da atividade cerebral de fundo e persistência de paroxismos focais após injeção de diazepam no diagnóstico de lesões cerebrais focais, potencialmente ressecáveis. (5) Chugani com auxílio de PET scan tem descrito a identificação de áreas displásicas com boa evolução das crises após procedimento de ressecção da área acometida. (12,70)
Do ponto de vista etiológico a síndrome de West pode ser dividida em 2 subgrupos: criptogênico ou provavelmente sintomático, mas de etiologia ainda desconhecida, e sintomático com etiologia determinada, incluindo afecções pré e peri-natais, síndromes neuro-cutâneas (especialmente esclerose tuberosa), malformações cerebrais, infecções cerebrais e distúrbios metabólicos (como a fenilcetonúria). É ainda discutível a existência de West idiopático e de casos precipitados por resposta imunomediada (pós- vacinal)(18,19,30,39)
A corticoterapia com o ACTH é o tratamento medicamentoso de primeira escolha na grande maioria dos casos inclusive naqueles sabidamente sintomáticos. Deve-se no entanto, sempre aplicar a regra do risco-benefício. Nos pacientes com encefalopatia grave, desnutridos com maior potencial para desenvolver infecções pulmonares deve-se evitar o uso do ACTH. A dose preconizada é de 30-150 u/m2 de superfície corpórea durante um período mínimo de 4 a 6 semanas. O controle da pressão arterial,do peso e dos distúrbios metabólicos é imprescindível devendo-se ser cuidadosamente monitorizadas. A utilização concomitante e mantida de Ácido Valproico na dose de 30-60 mg/Kg/dia (3 tomadas) é recomendada.(5) Muitos casos beneficiam-se do uso concomitante de benzodiazepínicos. Damos preferência ao clobazan na dose de 0,5 a 1 mg/Kg/dia (3 tomadas) devido a menor frequência de sedação, hipersecreção brônquica e melhor tolerabilidade. O uso de altas doses de piridoxina, TRH, imunoglobulina tem sido relatado em alguns trabalhos não sistematizados e o resultado satisfatório varia muito nas diversas séries da literatura.
Ressaltamos que nos casos de S. West que acompanham a esclerose tuberosa ou refratários ao ACTH deve-se lembrar o uso da Vigabatrina (45-150 mg/Kg/dia) sendo preconizada como de primeira escolha em muitas séries. (61,72)
Em relação ao tratamento cirúrgico a detecção precoce de anormalidades localizadas ou lateralizadas, correspondendo a áreas de hipometabolismo, detectados no PET de pacientes com West criptogênico vem sendo evidenciadas, sugerindo que uma lesão cerebral focal nessa fase da vida possa ter um papel importante na gênese dos espasmos.(12,70)
Alguns trabalhos mostram excelentes resultados após ressecção cirúrgica dessas lesões e as indicações cirúrgicas incluem: ausência de respostas ao ACTH e outras DAE e evidência de área de anormalidade cortical identificada pela convergência de achados de exames estruturais e funcionais que citaremos abaixo. As contra-indicações à cirurgia são a presença de doenças degenerativas ou de depósito e risco de seqüelas cirúrgicas inaceitáveis. Eventuais outras contra-indicações são as determinadas pelas condições clínicas do paciente. (39)
O tratamento cirúrgico quando indicado deve ser realizado precocemente, evitando-se assim maiores danos sobre o desenvolvimento.(39)
Os exames complementares para investigação etiológica da síndrome e investigação pré-cirurgica incluem: vide-eletroencefalograma; exames de imagem (RM e PET); potencial evocado somatossensitivo que avalia assimetrias das vias tálamo-corticais (latência e amplitude) e que pode providenciar dados localizatórios em alguns casos cirúrgicos, permitindo também que se avalie a integridade funcional do hemisfério oposto ao lado da cirurgia; a eletrocorticografia intra-operatória pode segundo alguns autores auxiliar a orientação da extensão da ressecção a ser realizada em bases neurofisiológicas. (12,70)
Epilepsia Mioclônica Grave – Síndrome de Dravet
A epilepsia mioclônica grave recentemente denominada de síndrome de Dravet é uma das apresentações sindrômicas mais refratárias de epilepsia da criança. A etiologia não é conhecida e tem discreto predomínio no sexo masculino com antecedente familiar frequente para epilepsia e convulsão febril. De aparecimento precoce, dentro do primeiro ano de vida, caracteriza-se inicialmente pelo aparecimento de crises febris, do tipo clônica ou unilaterais, de duração prolongada e de recorrência periódica (geralmente a cada 15 a 30 dias) acometendo uma criança previamente sem anormalidades neurológicas.(26,56) A hipertermia associada as crises costuma ser discreta e raramente excede os 380 C. Crises mioclônicas podem ocorrer entre o primeiro e quarto anos de vida, com quedas da cabeça e também mioclonias segmentares muitas vezes desencadeadas pelas variações da intensidade luminosa. Algumas crianças apresentam neste período outros tipos de crises incluindo as crises de ausência atípica de difícil distinção das crises parciais complexas associadas a automatismos e fenômenos autonômicos. O estado de mal epiléptico pode também aparecer neste período. Com o início das crises mioclônicas, ocorre atraso no desenvolvimento neurológico global, com gradual aparecimento de sinais neurológicos como ataxia, hiperreflexia e desenvolvimento de hiperatividade ou distúrbio pervasivo do desenvolvimento (padrão atípico de autismo/psicose). O EEG inicialmente é normal ou apresenta alentecimento da atividade elétrica cerebral de base, difuso ou unilateral. Após o aparecimento das crises mioclônicas o EEG modifica-se mostrando CPO e PPO generalizados, irregulares, de freqüência variável, que aumentam com a sonolência e são ativados pela fotoestimulação intermitente. Pode ocorrer resposta fotoparoxística em alguns casos. A anormalidades focais ou multifocais podem estar associadas. Pode ocorrer também no início da epilepsia atividade teta-ritmica de 4 a 5 Hz nas áreas centro-parietais e vértex. (17)
Em relação ao tratamento, as tentativas terapêuticas mostram resultado muito decepcionantes. As crises epilépticas, independentemente do regime de DAEs utilizado são sempre de difícil controle medicamentoso . Atualmente são descritos relatos esporádicos de resultado satisfatório com o uso dos brometos, do estiropentol e melhora das crises com o uso de melatonina. A lamotrigina deve ser evitada na síndrome de Dravet uma vez que pode determinar aumento na frequência das crises epilépticas ou mesmo induzir a estado de mal.(37,55,59)
Síndrome De Lennox-Gastaut
A síndrome de Lennox-Gastaut é uma forma grave de epilepsia, caracterizada por tríade caracterizada por polimorfismo de crises epilépticas recorrentes (principalmente crises tônicas, atônicas e ausências atípicas), retardo ou involução neuropsicomotora e um padrão eletrencefalográfico característico, constituído por alentecimento da atividade de base, presença de complexos lentos ponta-onda (1,5 a 2,5 Hz) de projeção difusa e predomínio anterior e descargas de polipontas (trem de espículas) de projeção difusa no registro eletrencefalográfico realizado em sono Fig 3 a e b (17,32). A síndrome de Lennox Gastaut é responsável por 2 a 3% das epilepsias da infância (8). Ocorre em geral em crianças pré-escolares de 1 a 7 anos de idade, com pico entre os 3 a 5 anos. A apresentação precoce antes dos 2 anos de vida pode seguir a síndrome de West e casos de início tardio, após os 10 anos de vida são mais esporádicos.(8,29) Antecedentes familiares para epilepsia variam muito nas diversas séries da literatura com cifras de 3 a 40%.
Em relação a Classificação etiológica grande parte dos casos, os denominados sintomáticos, ocorrem em crianças com encefalopatia prévia secundária a várias causas como encefalopatia hipóxico-isquêmica, displasias cerebrais, neurofacomatoses entre outras. (8,29) Entretanto, numa proporção variável entre 20 a 30% dos casos a etiologia não é estabelecida. A proposta recente da ILAE para estes casos denominados previamente denominados de criptogênicos e que sejam classificados como provavelmente sintomáticos uma vez que a classificação etiológica pode ser sugerida a medida que aumenta-se o grau de sofisticação dos recursos de neuroimagem funcional.(17,18,19)
As crises epilépticas mais comuns na síndrome de Lennox-Gastaut são as crises tônicas, atônicas e ausências atípicas, mas outros tipos de crises epilépticas podem estar associadas, tais como crises mioclônicas, crises parciais e crises generalizadas tônico-clônicas.(19) As crises de queda ao solo (drop attacks) são comuns e são mais frequentemente crises tônicas do que crises atônicas ou mioclônicas.
Fig 3. Padrão Eletrencefalográfico na Síndrome de Lennox- Gastaut
(a) 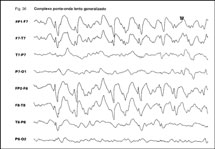 (b)
(b) 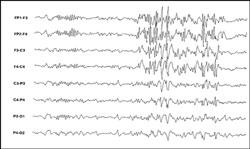
a) Complexo lento ponta-onda b) polipontas (trens de espículas)
Fig 3a. Complexo lento ponta-onda 3 b) Polipontas (trem de espículas)
Os mecanismos etiopatogênicos da síndrome de Lennox-Gastaut permanecem totalmente desconhecidos. Processos comuns que envolvem as epilepsias generalizadas idiopáticas podem estar envolvidos, colocando o perfil sindrômico da SLG dentro de um contínum biológico entre as epilepsias generalizadas idiopáticas e sintomáticas, na qual interagem fatores genéticos, neuromodulação, neuroplasticidade e fatores ambientais múltiplos determinando graus variados de expressão clínica e gravidade da encefalopatia epiléptica. (25,68)
Variações da expressão clínica idade-dependente
A síndrome de Lennox-Gastaut aparece antes dos dois anos de idade em duas circunstâncias: como “caso novo” ou após a síndrome de West. Nos casos de início precoce os traçados são inespecíficos com raros complexos ponta-onda lentos entretanto ele torna-se característicos com a idade de 2 a 3 anos. Nos casos em que a síndrome de Lennox-Gastaut segue a síndrome de West observa-se um período de transição de difícil diferenciação da síndrome de West e da síndrome de Lennox-Gastaut sendo que estas síndromes geralmente coexistem, em alguns casos um padrão hipsarritmico permanece por vários anos.(8,17,29)
Em adolescentes a síndrome de Lennox-Gastaut pode aparecer em 3 circunstâncias: “de novo”, após uma epilepsia parcial ou após epilepsia generalizada idiopática., entretanto em muitos casos o quadro eletroclínico é incompleto e transitório. Em vários pacientes dessa faixa etária a deterioração mental foi extremamente grave, a sintomatologia e a resposta ao uso de DAEs foram diferentes de acordo com a idade de início. Quando as crises iniciavam-se com a idade de 13 e 15 anos a síndrome foi completa e com pior prognóstico. Quando as crises epilépticas iniciavam-se com a idade de 16 a 19 anos a síndrome tem apresentação incompleta. Tanto as crises tônicas noturnas como algumas vezes os drop attacks podem estar ausentes nestes casos, mas as crises tônico-clônicas são geralmente muito frequentes. (8,29)
Há diferentes mudanças na semiologia de acordo com a faixa etária, por exemplo as crises de queda da cabeça, comuns na criança pequena tornam-se espasmos tônico ou desaparecem no decurso dos anos. Crises tônicas as quais geralmente são breves e freqüentes em crianças jovens tornam-se mais “fortes” e duradouras em crianças mais velhas. Os componentes clônicos e a assimetria na distribuição das crises começam a ser mais freqüentes e os adolescentes iniciam com as crises generalizadas tônico-clônicas e com as crises do tipo vibratórias em que somam-se às clássicas crises tônicas. As ausências atípicas como também as crises mioclônicas tendem a diminuir com a idade e os drop attacks persistem principalmente devido a contração tônica do que à atonia. As crises noturnas tônicas persistem mesmo quando as crises diurnas tornam-se mais raras. O padrão eletrencefalográfico típivo de complexo lento ponta-onda tende a diminuir gradualmente sendo substituído por ondas lentas de baixa frequência e baixa voltagem. Por outro lado as polipontas (o trem de espículas) persiste durante o sono. Com a evolução, os casos de síndrome de Lennox-Gastaut sintomático com grave retardo mental podem evoluir para um padrão eletrencefalográfico de espículas independentes e múltiplas denominadas por vários autores de epilepsia grave com múltiplos focos de espículas independentes (8,17,58). Nestes casos embora as ausências atípicas não sejam mais observadas as crises tônicas e os drop attacks mantém-se freqüentes.
A grande maioria dos pacientes com síndrome de Lennox-Gastaut tem retardo mental, de moderado a grave, mas eventualmente algumas crianças podem apresentar um desenvolvimento normal, principalmente se as crises são precocemente controladas. No entanto, é importante ressaltar que alguns casos o retardo mental pode ocorrer mesmo antes do aparecimento das crises epilépticas ou progride mesmo quando as crises epilépticas são controladas. Alguns pacientes com apresentação de início tardio podem ter preservação cognitiva. (25)
A resposta clínica da SLG às várias DAEs disponíveis é habitualmente bastante reservada, e grande parte dos casos mantém crises frequentes mesmo em uso de regime politerápico.
As medicações que parecem mais efetivas, ao nosso ver, são o valproato, associado a benzodiazepínicos (principalmente clobazan), a lamotrigina e o topiramato nos casos de crises de drop attacks. Em nossa experiência a calosotomia auxiliou apenas transitoriamente os episódios de queda cefálica. Sua indicação portanto reserva-se aos casos com traumatismo crainioencefálico freqüente ou potencial. É importante ressaltar que o curso clínico da SLG é bastante variável, no que se refere a oscilações da frequência e intensidade das crises epilépticas, e muitas vezes a flutuação não depende nem mesmo da medicação ou do regime de DAEs empregado. A dieta cetogênica tem sido uma opção válida principalmente no controle das crises tônicas e ausências atípicas bem como na melhora cognitiva de muitos pacientes com crises refratárias.(32,39,40,47)
Síndrome de Doose
Também conhecida como epilepsia mioclônico-astática, ocorre em 0,2% das crianças com epilepsia, tem nítido predomínio nos meninos. A história familiar positiva tanto para convulsão febril e crises afebris expressam a forte predisposição genética da síndrome. O quadro clínico caracteriza-se por crises predominantemente generalizadas com crises mioclônicas, astáticas e mioclônico-astáticas. A epilepsia inicia-se dos 2 até 5 anos de idade e as crises astáticas características constituem-se em episódios de perda do tonus postural com quedas, algumas vezes precedidas por crises mioclônicas segmentares dos membros superiores e da face.(24,58) O Eletrencefalograma pode apresentar atividade de base lenta constituída por ritmos teta de 4 a 7 Hz de predomínio em áreas parietais. A atividade paroxística varia de acordo com a crises epiléptica envolvida predominando os paroxismos generalizados de complexo ponta-onda 2 a 3 Hz, e os complexos poliponta-onda.(17) Embora o quadro clínico pode ser de difícil diferenciação com a síndrome de Lennox- Gastaut ressalta-se a forte predisposição genética e o predomínio de crises mioclônicas na síndrome de Doose. As crises tônicas características da SLG, são mais raras ou acompanham os casos de evolução desfavorável.(24) O prognóstico da Síndrome de Doose é variável. Alguns casos tem remissão completa sem anormalidades neuropsicológicas residuais. Outros evoluem com deterioração cognitiva, crises de difícil controle, ataxia e hiperatividade. O pior prognóstico relaciona-se ao início antes do 1 ano de vida e a presença de estado de mal epiléptico.
O Ácido Valpróico, em monoterapia ou associado a benzodiazepínicos parece ser a estratégia terapêutica de melhor resultado. Casos resistentes podem ter resposta variável ao ACTH, felbamato bem como a dieta cetogênica. (42,51)
Epilepsia com ausência mioclônica
Epilepsia caracterizada pela recorrência de crises de ausência mioclônicas difusas, bilaterais, simétricas, com duração em média de 10 a 60 segundos, de intensidade variável por vezes pode ser acompanhada por um componente tônico que muitas vezes pode ser assimétrico levando a uma versão lateral ou ântero-posterior do corpo e da cabeça. A contração tônica é máxima em ombros e músculo deltóide e é responsável pela elevação dos braços. Os movimentos mioclônicos envolvem principalmente braços, ombros e pernas, os músculos da face são menos frequentemente envolvidos e quando ocorre as mioclonias faciais geralmente envolvem a boca e as manifestações autonômicas como mudança do padrão respiratório e perda urinária podem ser observados junto com as mioclonias.(1,26,27,37,55) Há um predomínio no sexo masculino. O início da epilepsia ocorre em média aos 7 anos de idade (média de 2 a 12 anos).(30,48,49) A história familiar de epilepsia é encontrada em 25% dos casos. O EEG mostra complexos ponta-onda de 3 Hz e há uma estrita correlação entre o início da descarga e a mioclonia. Pode estar associada a outros tipos de crises epilépticas em 63% dos casos, como as crises generalizadas tônico-clônicas, ausências e por vezes, menos frequentemente o estado de malmioclônico.(17) A evolução é variável, podendo ocorrer um comprometimento intelectual antes ou no início do aparecimento das crises de ausência mioclônica, em 50% dos casos as crises epilépticas são refratárias às várias drogas antiepilépticas em combinação. (1,15)A associação medicamentosa com melhores resultados é o valproato, etossuximida e lamotrigine.(1,3,23,52).
Epilepsias catastróficas da Infância
Há um grupo de síndromes epilépticas graves, que , salientando-se a refratariedade, o impacto neuropsicológico e a intratabilidade com as várias DAE foram denominadas de epilepsias “catastróficas”.(68) Tais quadros ressaltam-se não só pelas crises epilépticas de difícil controle medicamentoso mas sobretudo sobre o atraso do desenvolvimento neuropsicomotor ou mesmo regressão das funções cognitivas. Entre elas temos: síndrome de Ohtahara, epilepsia mioclônica severa, síndrome de West, síndrome de Lennox-Gastaut, epilepsia com crises mioclônico astáticas, síndrome de Sturge Weber e encefalite de Rasmussen. Algumas já foram descritas acima e outras serão abordadas a seguir, entre elas:
Síndrome de Sturge-Weber
A síndrome de Sturge-Weber (SSW) foi descrita no final do século passado por Schirmer, porém foi em 1879 que Sturge a caracterizou clinicamente. A SWW é uma neurofacomatose de origem mesodérmica caracterizada por nevo vascular facial unilateral e angioma em leptomeninge ipsilateral, que leva a uma atrofia progressiva focal hemisférica com hemiparesia. O angioma facial é congênito, geralmente unilateral, e localiza-se em uma ou mais das três divisões faciais do nervo trigêmeo, podendo acometer também a nasofaringe e a membrana coroidal ocular, o que resulta em glaucoma em 25% dos casos. Pode-se estender para outras partes da face, incluindo lábios, palato, língua, faringe e laringe, assim como para tronco e membros, ipsi ou contralateralmente. Os sinais e sintomas neurológicos dos pacientes são decorrentes da presença do angioma venoso leptomeníngeo. O início frequentemente é precoce com crises parciais motoras, e também pode ocorrer crises secundariamente generalizadas, mioclônicas, atônicas e espasmos infantis. A paralisia de Todd (paralisia pós crítica) pode aparecer pela grande freqüência das crises epilépticas.
As crises epilépticas podem tornar-se progressivamente refratárias ao tratamento medicamentoso e em 30% dos casos são acompanhadas por hemiparesia progressiva. A hemiparesia pode ser observada em 25% a 50% dos casos, podendo ser bilateral em pacientes com angiomas bilateral. Desempenho mental subnormal ocorre em 50 a 75% dos casos. As calcificações cerebrais ocorrem no córtex cerebral subjacente à angiomatose pial e são mais comumente vistas nas camadas 2 e 3 do córtex cerebral, de topografia parietal e parieto-occipital. O hemisfério cerebral ipsilateral é geralmente atrófico. Se não houver controle adequado das crises epilépticas com o tratamento medicamentoso, o tratamento cirúrgico é indicado.
Exames de tomografia de crânio e ressonância nuclear magnética cerebral repetidos ao longo do tratamento podem revelar aumento das lesões calcificadas, assim como demonstrar a atrofia córtico-subcortical, realçando o caráter progressivo da doença. (12,70,71). Nestes casos a hemisferectomia tem sido preconizado por alguns autores Fig 4.
Fig 4. Calcificação Giral na S. Sturge-Weber
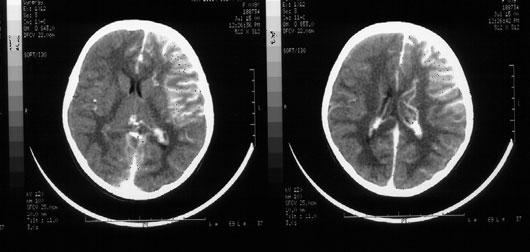
Encefalite Crônica de Rasmussen
O primeiro sinal de encefalite crônica de Rasmussen é o aparecimento de crises epilépticas parciais com ou sem generalização subsequente. Em 50% dos casos a epilepsia parcial contínua ocorre em poucos anos após o início sendo que o estado de mal convulsivo ocorre em 20 % dos pacientes. Nos estágios iniciais da doença, o diagnóstico de encefalite crônica pode não ser percebido, a suspeita é realizada com o desenvolvimento de deterioração neurológica progressiva, começando no primeiro ano em 40% dos pacientes, no segundo e terceiro ano em 40 % dos pacientes e em 4 a 15 anos do início em 20% dos pacientes. O primeiro sinal que aparece é uma hemiparesia lenta e progressiva pode associar-se a defeitos no campo visual. A deterioração intelectual e distúrbio de comportamento somam-se a sintomaltologia inicial.. A investigação neuroradiológica mostra uma atrofia cerebral unilateral progressiva com início na região têmporo-insular, que pode ser observada na tomografia de crânio e na ressonância nuclear magnética, o SPECT cerebral mostra uma área de hipoperfusão correspondente a área de hemiatrofia hemisférica (12,70,71) O EEG mostra frequentemente alteração na atividade elétrica cerebral de base bilateral, as anormalidades tendem a ser mais no entanto assimétricas envolvendo o hemisfério acometido, as alterações mais frequentes envolvem ondas lentas generalizadas principalmente no hemisfério lesado e espículas focais menos proeminentes.(17)
As alterações patológicas observadas na síndrome de Rasmussen não são específicas e as características clínicas variam de acordo com a duração e atividade da doença. Nos estágios iniciais, a encefalite crônica é caracterizada por nódulos microgliais, linfócitos e monócitos na bainha perivascular, espongiose também pode ser observada associada a processos inflamatórios.
Possíveis mecanismos etiológicos podem ser identificados como: infecção viral crônica, infecção viral aguda com resposta imune local e processo auto-imune independente não relacionado à infecção conhecida. As crises epilépticas são refratárias com drogas antiepilépticas habituais, geralmente necessita politerapia com altas doses de drogas antiepilépticas. A hemisferectomia é mais efetiva no controle das crises epilépticas. Entre outras tentativas medicamentosas temos o uso de esteróides, gamaglobulina intravenosa e terapia antiviral (interferon, acyclovir e zidovudine) sendo que a escolha reflete o mecanismo causal presumido.(3,4,17)
Síndrome de Landau e Kleffner
Embora a epilepsia associado a síndrome de Landau-Kleffner pode apresentar-se com bom prognóstico, o impacto neropsicológico da afasia é suficiente para colocar a síndrome no contexto de epilepsias graves. Essa síndrome, descrita pela primeira vez por Landau & Kleffner em 1957 em 6 crianças que desenvolveram afasia, após terem adquirido a linguagem normal associa-se a crises epilépticas generalizadas ou parciais com resposta variávelao uso de DAE sendo descritos casos até com remissão espontânea das crises. Em todos os pacientes o eletroencefalograma (EEG) mostrava-se anormal com descargas epileptiformes bilaterais síncronas de espícula-onda, predominantemente nas regiões temporais, estas anormalidades tendem a desaparecer com a evolução da síndrome (17) Não há evidência de lesão estrutural no cérebro.(12,70,71).Observou-se que a afasia, mais propriamente uma agnosia auditiva, tem uma evolução bastante variável, havendo relatos de casos com boa evolução e outros com prognóstico reservado.
O início da síndrome ocorre por volta de 4 a 7 anos de idade, fase em que a maior parte das funções neurológicas está em formação ou em maturação. A literatura atual ainda não mostra consenso sobre os mecanismos que levariam à epilepsia e afasia, assim como os distúrbios de comportamento na SLK dando-se ênfase aos mecanismos de desconexão de áreas cerebrais que estão intimamente relacionadas a linguagem.
O uso de corticsteróide e de gamaglobulina hiperimune tem sido referido por alguns autores como efetivo tanto para o controle das crises como para a melhora da própria afasia. A transecção subpial múltipla, preconizada como efetiva por Morrell, tem sido bastante contestada em séries recentes.
Tratamento clínico da epilepsia refratária na infância
Grande parte das epilepsias refratárias ocorrem dentro do contexto de franca encefalopatia, algumas de prognóstico bastante reservado em relação a possibilidade de controle das crises, mesmo se considerados o tratamento cirúrgico, a dieta cetogênica e as novas DAEs.(7,39) Assim, em muitos casos, a freqüência de crises nesses pacientes com crises epilépticas de difícil controle medicamentoso pode ser diminuídas pela medicação mas não completamente controlada Portanto a ênfase na abordagem terapêutica centra-se no conceito de risco-benefício e na utilização planejada de associações de DAEs denominada por alguns autores de “politerapia racional”. (3,10,14,15,32,52)
Nas encefalopatias epilépticas é importante considerar o risco de indução de estado de mal epiléptico pela associação de drogas potencialmente sedativas. (10,37,50)
Na síndrome de Lennox-Gastaut, a sonolência induzida por uso excessivo de benzodiazepínicos para controle das crises deteriora o quadro clinico do paciente, podendo levar a um status epilepticus tônico ou confusional.(32,40) Por isso, achamos necessário nesse tipo de encefalopatia epiléptica reduzir a ansiedade dos médicos diante de crises que, apesar dos esforços terapêuticos, não serão totalmente controladas na grande maioria dos casos, e que incrementos exagerados das doses das drogas antiepilépticas causam mais danos (alterações cognitivas, sedação extrema, intoxicação), do que benefícios clínicos mensuráveis. No tratamento farmaco1ógico das epilepsias devem ser seguidos alguns critérios como: diagnóstico preciso e atual das crises epilépticas, pesquisa de fatores desencadeantes das crises epilépticas; fortalecimento da adesão ao tratamento proposto e escolha adequada das drogas antiepilépticas de efeito potencialmente sinérgicos. (3,10,61)
O Ácido Valpróico é considerado a medicação de eleição nas encefalopatias epilépticas da infância uma vez que estas associam-se a um polimorfismo de crises que incluem ausências, crises atônicas que podem ser exacerbadas pelo uso de drogas como o fenobarbital, a carbamazepina e a fenitoína. Nestes casos dá-se recomenda-se a associação de ácido valpróico com um benzodiazepínico, dando-se preferência para aquele de menor efeito sedativo e menor risco de hipersecreção brônquica como é o caso do clobazam. Ao prescrevermos o fracionamento da dose é fundamental respeitarmos a vida média de cada medicação antiepiléptica sendo que os ajustes das doses das DAE devem ser realizados após o tempo necessário para que seja obtido um platô estável de concentração plasmática da droga. Em geral, o estado de equilíbrio demora o equivalente a 5 meias-vidas para se estabelecer.(3,14,32,50,52) Assim este mesmo tempo é necessário quando se programa uma nova associação medicamentosa.
Tabela 1
| TRATAMENTO FARMACOLÓGICO AS EPILEPSIAS NA INFÂNCIA | ||
|
Tipos de Crises |
1ª escolha |
2ª escolha |
|
Parcial |
Carbamazepina |
Valproato (Depakene) 20-60 mg/kg/dia |
|
Mioclônias |
Valproato |
Lamotrigina (5-8 mg/Kg/dia) |
|
Espasmos infantis |
ACTH |
Vigabatrina |
|
Ausências |
Valproato |
Etossuximida |
|
Tônico-clônica |
Valproato |
Carbamazepina |
Lembramos o fenômeno de "saturação cinética", que ocorre com a fenitoína e indução enzimática com a carbamazepina. No caso da fenitoína reflete-se pelo fato de que mesmo pequenos incrementos na dose produzem grandes aumentos nos níveis séricos podendo levar a quadros de intoxicação. No caso da auto-indução que ocorre com a carbamazepina os níveis séricos podem cair transitóriamente devido a autometabolização da droga nas primeiras 3 a 6 semanas do seu uso inicial., não necessitando ajustes de dose nesta fase para não ocasionar futura intoxicação. (52)
Nos casos nos quais a politerapia está indicada é necessário conhecer as interações medicamentosas entre as várias DAE e seus potenciais efeitos favoráveis ou desfavoráveis (Tabela 3). Lembramos a associação entre drogas indutoras do metabolismo hepáticos como o fenobarbital com inibidoras como o ácido valpróico, determinando níveis elevados de fenobarbital , que podem levar o paciente a um estado de sonolência ou ataxia, devido a intoxicação barbitúrica. Deve-se evitar a associação de drogas potencialmente sedativas como o fenobarbital e a carbamazepina com os benzodiazepínicos. Drogas indutoras do metabolismo hepático com mecanismo de ação semelhante como a associação da carbamazepina com a fenitoína devem ser evitadas, .uma vez que além de não possuem efeitos sinérgicos podem ocasionar interações recíprocas com diminuição dos níveis séricos de ambas as drogas.(52) As novas drogas antiepilépticas desenhadas originalmente para ter um perfil amplo, poucos efeitos colaterais, baixa ligação protéica e menor interação medicamentosa ocupam apenas um pequeno espaço no controle das crises refratárias.(3,10,32,50) Sua utilização em nosso meio também é muitas vezes proibitiva pelo seu alto custo financeiro, principalmente quando avaliamos população de baixa renda. A Tabela (2) expõe o custo financeiro comparado entre as drogas habituais e as novas DAEs disponíveis no mercado brasileiro.
Das novas DAEs os maiores benefícios incluem o uso da Vigabatrina, no controle dos espasmos infantis associados a esclerose tuberosa, a lamotrigina para as crises generalizadas atônicas e mioclônicas (da síndrome de Lennox-Gastaut ou na síndrome de Rett), podendo potencialmente acentuar as crises mioclônicas em outras síndromes epilépticas como na síndrome de Dravet, a gabapentina como coadjuvante no tratamento de crises parciais refratárias e o topiramato para auxílio no controle de crises generalizadas atônicas, tônicas e ausências associadas a encefalopatias epilépticas graves.(4,23,35,44,67,77) O uso atual do Sulthiame (Ospolot), parece ter um valor na expressão eletrofisiológica das descargas epileptiformes associadas a crises parciais como na epilepsia parcial atípica ou nos casos de descargas contínuas associados ao estado de mal elétrico do sono lento ou na síndrome de Landau-Kleffner.
Tabela 2 – Custo das drogas antiepilépticas
| Drogas habituais |
Custo |
Novas drogas |
Custo |
|
Carbamazepina |
R$ 59 |
Vigabatrina |
R$260 |
|
Valproato |
R$45 |
Lamotrigina |
R$300 |
|
Fenobarbital |
R$ 10 |
Topiramato |
R$236 |
|
Fenitoína |
R$ 20 |
Oxcarbazepina |
R$150 |
|
|
|
Gabapentina |
R$246 |
A farmacocinética ideal para as drogas antiepilépticas inclui: alta disponibilidade oral, nenhuma ou baixa ligação protéica plasmática, meia vida longa, dose de uma ou duas vezes por dia, cinética linear, nenhum metabólito ativo, eliminação renal, nenhuma indução enzimática, nenhuma interação medicamentosa.
Dentre as medicações de uso alternativo para epilepsia de difícil controle temos o piracetam que tem melhor efeito sobre as mioclonias de origem cortical e nas mioclonias associadas a epilepsia mioclônica progressiva e também uma melhora sobre a freqüência de convulsões. O levetiracetam parece ser uma droga antiepiléptica promissora no controle de crises associadas a fotossensibilidade.(44,47)
Tabela 3
|
Substância Ativa |
Indicações |
Produto |
Posologia |
Meia-vida |
Número |
Observações |
|
Fenobarbital |
CGTC |
Gardenal |
Cr=3-5mg/kg |
Cr=37-73 |
1-2 |
Sonolência no início do tratamento, risco de hiperexcitabilidade paradoxal (criança e idosos) |
|
Carbamazepina |
CPS-CPC |
Tegretol |
Cr=10-30mg/kg |
Cr=9-19 |
2-4 |
É comum leucopenia |
|
Oxcarbamazepina |
CGTC |
TrileptalÒ |
Cr=10-30mg/kg |
9-10 |
2-3 |
Cansaço, vertigem, sonolência |
|
Fenitoína |
CGTC |
Hidantal |
Cr=5-6mg/kg |
Cr=5-14 |
1-3 |
Hiperplasia gengival |
|
Ácido valpróico |
CGA |
Depakene |
Cr=20-60mg/kg |
Cr=6-18 |
2-4 |
Distúrbios gastrointestinais no início do tratamento. Foram descritos casos de hepatoxicidade grave |
|
Clonazepan |
CGM |
Rivotril |
Cr=0,1-0,2mg/kg |
Cr=20-40 |
2-3 |
Sonolência excessiva, principalmente com doses elevadas |
|
Lamotrigina |
CGM |
LamictalÒ |
Cr=2-8mg/kg |
24-41 |
2 |
Sonolência, rash cutâneo, diplopia, náuseas, ataxia e vômitos |
|
Topiramato |
CGM |
TopamaxÒ |
Cr=2-10mg/kg |
24-41 |
2 |
Sonolência, perda de peso, calculo renal, diarréia, náuseas, ataxia e vômitos |
|
Vigabatrina |
Espasmos |
Sabrilâ |
Cr= 50 a |
5-8 |
2 |
Agitação psicomotora |
|
Etosuximida |
CGA |
Zarontin |
Cr=10-20mg/kg |
Cr=20-60 |
2-3 |
Algumas vezes é necessário usar em conjunto com outras drogas devido ao risco de convulsões |
1 - Citamos apenas alguns nomes a título de exemplo. Outros produtos e suas apresentações podem ser encontrados nos textos apropriados.
2 - As doses terapêuticas indicadas devem ser consideradas apenas como guia. Alguns pacientes não tolerarão nem a dose mínima e outros poderão necessitar de doses mais elevadas para atingir um nível terapêutico efetivo.
3 - Os dados citados podem sofrer alterações quando as drogas são ministradas em associação com outras drogas, anti-epilépticas ou não.
4 - Quando fracionada, a dose diária total deve ser administrada em doses iguais, em intervalos regulares, por exemplo de 8/8 h.
5- Não foi colocada a vigabatrina na tabela, devido à relatos recentes de restrição do campo visual associado ao seu uso.
CGTC=crise generalizada tônico-clônica (convulsão generalizada)
Cr = crianças CPS = crise parcial simples CGM= crise generalizada mioclônica + = média
0 = praticamente desconsiderável CPC = crise parcial complexa CGA= crise generalizada ausência ++ = intensa
Dieta Cetogênica
As manipulações dietéticas e o jejum como forma de tratamento tem sido utilizados há centenas de anos. (42,43)
Wilder propôs em 1921 que uma dieta rica em gorduras e pobre em carboidratos e proteínas em que poderia reproduzir a cetose e a acidose metabólica características do jejum, além de permitir a manutenção deste estado por período de tempo Essa dieta recebeu a denominação de dieta cetogênica.
O uso da dieta cetogênica no controle de pacientes epilépticos teve destaque durante a década de 20, quando apenas duas DAE eram conhecidas, os brometos e o fenobarbital. Com o advento de novas e potentes drogas nas décadas seguintes, a dieta cetogênica praticamente deixou de ser usada em quase todos os centros médicos.(72)
A dieta cetogênica é indicada principalmente para crianças epilépticas com mais de um ano de idade, que tenham epilepsia comprovadamente refratária a duas ou mais drogas utilizadas em doses adequadas ou, então para os pacientes cujos efeitos adversos das DAE sejam intoleráveis ou que tenham reações idiossincrásicas a elas. A dieta cetogênica parece atuar melhor em crises generalizadas (crises de ausência e mioclônicas) e em epilepsias como a síndrome de Lennox-Gastaut e síndrome de West, porém qualquer tipo de crises pode beneficiar-se com essa modalidade terapêutica. (6,7,42,43)
O mecanismo de ação da dieta cetogênica no controle das crises epilépticas ainda não é bem conhecido. Existem várias teorias para explicá-la, destacando-se a cetose produzida pelos corpos cetônicos e a acidose que a acompanha, além das alterações no balanço de eletrólitos, de fluídos e de alterações na concentração de lipídios e adaptações metabólicas induzidas pela dieta no cérebro. Esses mesmos autores atribuíram o efeito antiepiléptico à cetose e ao efeito sedativo do ácido acetoacético. A cetose e a acidose decorrentes da catabolização cerebral de gorduras certamente diminui a excitabilidade neuronal e eleva o limiar convulsivógeno. O efeito anticonvulsivante da dieta cetogênica deve estar relacionado a outros fatores metabólicos envolvidos e pouco conhecidos.(6,7)
Realizamos um trabalho com o objetivo de analisar os fatores preditivos clínicos e eletrencefalográficos de prognóstico em relação ao uso da dieta cetogênica em 10 crianças com epilepsia de difícil controle medicamentoso.
Em todos os pacientes houve melhora importante da qualidade de vida, os pacientes ficaram mais ativos, com menor sonolência e com melhora da disposição para as atividades habituais. Observamos uma boa aceitação da dieta, com exceção de uma paciente, em que foi retirada da dieta. Dois pacientes tiveram controle total das crises epilépticas, em 4 pacientes houve uma melhora de 50% e em 3 pacientes houve uma diminuição da frequência das crises em 30%. Não houve efeitos colaterais ou alterações metabólicas em nossos pacientes. A dieta cetogênica torna-se uma alternativa válida em pacientes que não responderam às medicações convencionais. Porém o sucesso dessa forma de tratamento está muito ligado à motivação dos familiares do paciente e à boa relação com uma equipe multidisciplinar composta por nutricionista, neurologista, psicólogo e assistente social.
Gráfico 1:

Deve-se ressaltar que o sucesso no tratamento de pacientes epilépticos não se resume nos conhecimentos dos aspectos biológicos das síndromes epilépticas . É muito difícil a aceitação de qualquer doença crônica e de sua limitações. Em particular, é muito difícil para a criança e sua família se adaptarem não somente à epilepsia, como também a seus riscos, complicações e eventuais atitudes hostis da sociedade.
Daí a necessidade de abordar o tratamento de uma forma multidisciplinar, sempre que possível, por uma equipe formada de médicos, enfermeiras, psicólogas e assistentes sociais, no intuito de dar um apoio conjunto ao paciente e à sua família, facilitar a aderência ao tratamento crônico, detectar precocemente os déficits neuropsicológicos específicos, orientar as medidas de reabilitação e acima de tudo auxiliar as mudança necessárias de atitudes hostis, de não aceitação e do preconceito diante das epilepsias de difícil controle.
Referências
1) AICARDI J. - Early myoclonic encephalopathy. In: Epileptic Syndromes in Infancy, Childhood and Adolescense. Ed: Roger J.; Dravet C.; Bureau M.; Dreifuss F.E.; Wolf P. London: John Libbey. 1985, Cap. 2, p. 12-22.
2) Aicardi J. Epileptic encephalopathies of early childhood. Curr Opin Neurol Neurosurg 1992 Jun;5(3):344-8.
3) Aicardi J. Risks and benefits of new antiepileptic agents in children. Rev Neurol 2000 Aug; 16-31; 31(4):376-81.
4) Akman CI, Schubert R. The role of tiagabine in the treatment of intractable epilepsy of childhood with multifocal independent spikes: a case report. Clin Electroencephalogr 2000 Oct; 31(4): 207-10.
5) Antoniuk SA, Bruck I, Spessatto A, Halick SM, de Bruyn LR, Meister E, de Paola D. West syndrome: clinical and electroencephalographic follow up of 70 patients and response to its treatment with adrenocorticotropic hormone, prednisone, vigabatrin, nitrazepam and valproate. Arq Neuropsiquiatr 2000 Sep; 58(3A):683-90.
6) BALLABAN-GIL K.; CALLAHAN C.: O’DELL C., PAPPO M.; MOSHÉ S.; SHINNAR S. - Complications of the ketogenic diet. Epilepsia, 39(7): 744-748, 1998.
7) BARRON T.F. & HUNT S.L. A review of the newer antiepileptic drugs and the ketogenic diet. Clin Pediatries, 36(9): 513-521, 1997.
8) BEAUMANOIR A. & DRAVET C. - The Lennox-Gastaut syndrome. In: Epileptic Syndromes in Infancy, Childhood and Adolescense. Eds: Roger J.; Dravet C.; Bureau M.; Dreifuss F.E.; Perret A.; Wolf P. London: John Libbey. 1992, p. 115-132.
9) BERG A.T.; LEVY S.R.; NOVOTNY E.J.; SHINNAR S. Predictors of intractable epilepsy in childhood: a case-control study. Epilepsia, 37(1): 24-30, 1996.
10) Bialer M.; Johannessen S.I.; Kupferberg H.J.; Levy R.H.; Loiseau P.; Perucca E. Progress report on new antiepileptic drugs: a summary of the Third Eilat Conference. Epilepsy res, 1996; 25(3): 299-319.
11) BOURGEOIS B.F.D. - General Concepts of Medical Intractability. In: Epilepsy Surgery. Ed. Lüders H.O. Raven Press, New York, cap.9, p. 77-81, 1991.
12) BRONEN R. A.; FULBRIGHT R. K.; SPENCER D. D.; SPENCER S. S.; KIM J. H.; LANGE R. C. - Refractory epilepsy: comparison of MR imaging, CT, and histopathologic findings in 117 patients. Radiology 201(1): 97-105, 1996.
13) Campistol J. Epileptic syndromes in the first year of life and congenital errors of metabolism]. Rev Neurol 2000 Jun; 30 Suppl 1:S60-74.
14) Campos Castello J. Therapeutic strategy in severe encephalopathies.Rev Neurol 2001 May 16; 32(9):860-866.
15) Chen Y.J., Kang W.M., Min So W.C. Comparison of Antiepileptic Drugs on Cognitive Function in Newly Diagnosed Epileptic Children: A Psychometric and Neurophysiological Study. Epilepsia; 1996 37(1): 81-86.
16) Chiron C, Marchand MC, Tran A, Rey E, d'Athis P, Vincent J, Dulac O, Pons G. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo-controlled syndrome-dedicated trial. STICLO study group. .Lancet 2000 Nov 11; 356(9242):1638-42.
17) COMMISSION ON CLASSIFICATION AND TERMINOLOGY OF THE INTERNATIONAL LEAGUE AGAINST EPILEPSY. - Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia, 22: 489-501, 1981.
18) COMMISSION ON CLASSIFICATION AND TERMINOLOGY OF INTERNATIONAL LEAGUE AGAINST EPILEPSY. - Proposal for classification of Epilepsies and Epileptic syndromes. Epilepsia, 26: 268-278, 1985.
19) COMMISSION ON CLASSIFICATION AND TERMINOLOGY OF THE INTERNATIONAL LEAGUE AGAINST EPILEPSY. -Proposal for revised classification of Epilepsies and Epileptic syndromes. Epilepsia, 30: 389-398, 1989.
20) COMISSION ON EPIDEMIOLOGY AND PROGNOSIS, INTERNATIONAL LEAGUE AGAINST EPILEPSY. Guidelines for Epidemiologic Studies on Epilepsy. Epilepsia, 34(4): 592-596, 1993.
21) COWAN L.D.; BODENSTEINER J.B.; LEVITON A.; DOHERTY L. - Prevalence of the epilepsies in children and adolescents. Epilepsia, 30(1): 94-106, 1989.
22) COWAN L.D. & HUDSON L.S. - The epidemiology and natural history of infantile spasms. J. child neurol., 6: 355-364, 1991.
23) Culy CR, Goa KL. Lamotrigine. A review of its use in childhood epilepsy.Paediatr Drugs 2000 Jul-Aug; 2(4):299-330.
24) Doose H. Myoclonic-astatic epilepsy. Epilepsy Res Suppl 1992; 6:163-8.
25) Doose H, Hahn A, Neubauer BA, Pistohl J, Stephani U. Atypical "benign" partial epilepsy of childhood or pseudo-lennox syndrome. Part II: family study.Neuropediatrics 2001 Feb; 32(1):9-13.
26) DRAVET C.; BUREAU M.; ROGER J. - Severe myoclonic epilepsy of infants. In: Epileptic Syndromes in Infancy, Childhood and Adolescense. Eds: Roger J.; Dravet C.; Bureau M.; Dreifuss F.E.; Wolf P. London: John Libbey. 1985, Cap. 7, p. 58-67.
27) Dravet C. Severe myoclonic epilepsy in infants and its related syndromes. Epilepsia 2000; 41(Suppl. 9): 7.
28) Dulac O. Epileptic encephalopathy. Epilepsia 2001; 42 Suppl 3:23-6.
29) Dulac O, N'Guyen T. The Lennox-Gastaut syndrome.Epilepsia 1993; 34 Suppl 7:S7-17.
30) ERIKSSON K.J. & KOIVIKKO M.J. Prevalence, classification, and severity of epilepsy and epileptic syndromes in children. Epilepsia, 38(12): 1275-1282, 1997.
31) Echenne B, Dulac O, Parayre-Chanez MJ, Chiron C, Taillebois L, Cognot C, Andary M, Clot J, Baldy-Moulinier M. Treatment of infantile spasms with intravenous gamma-globulins. Brain Dev 1991 Sep; 13(5):313-9
32) French JA. The role of new antiepileptic drugs. Am J Manag Care 2001 Jul; 7(7 Suppl): S209-14.
33) GASTAUT H.; ROGER J.; SOULAYROL R.; TASSINARI C.A.; RÉGIS H.; DRAVET C.; BERNARD R.; PINSARD N.; SAINT-JEAN M. Childhood epileptic encephalopathy with diffuse slow spike waves (otherwise known as “petit mal variant”) or Lennox Gastaut syndrome. Epilepsia, 7: 139-179, 1966.
34) Genton P. When antiepileptic drugs aggravate epilepsy. Brain Dev 2000 Mar; 22(2):75-80.
35) Glauser TA. Topiramate in the catastrophic epilepsies of childhood. J Child Neurol 2000; 15 Suppl 1:S14-21.
36) Goldsmith I. L.; Zupanc M. L.; Buchhalter J. R. Long –term seizure outcome in 74 patients with Lennox-Gastaut syndrome: Effects of incorporating MRI head imaging in defining the cryptogenic subgroup. Epilepsia 2000; 41(4): 395-299.
37) Guerrini R, Dravet C, Genton P, Belmonte A, Kaminska A, Dulac O. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia 1998 May; 39(5):508-12.
38) HAUSER W.A.; ANNEGERS J.F.; KURLAND L.T. - Prevalence of epilepsy in Rochester, Minnesota, 1940 - 1980. Epilepsia, 32: 429-445, 1991.
39) HOLMES G.L. - Surgery for intractable seizures in infancy and early childhood. Neurology, 43(Suppl. 5): S28-S35, 1993.
40) Hosain S, Nikalov B, Harden C, Li M, Fraser R, Labar D. Vagus nerve stimulation treatment for Lennox-Gastaut syndrome. J Child Neurol 2000 Aug; 15(8):509-12.
41) Husain AM, Foley CM, Legido A, Chandler DA, Miles DK, Grover WD. West syndrome in tuberous sclerosis complex.Pediatr Neurol 2000 Sep; 23(3):233-5.
42) HUTTENLOCHER P.R. - Ketonemia and seizures: metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediat res. 10: 536-540, 1976.
43) HUTTENLOCHER P.R. & WILBOURN A. J. Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology, 21: 1097-1103, 1971.
44) Ikeda A.; Shibasaki H.; Tashiro K.; Mizuno Y.; Kimura J. Clinical trial of piracetam in patients with myoclonus: nationwide multiinstitution study in Japan. The Myoclonus/Piracetam study group. Mov disord, 1996; 11(6): 691-700.
45) Jan MM, Shaabat AO. Clobazam for the treatment of intractable childhood epilepsy. Saudi Med J 2000 Jul; 21(7): 622-4.
46) Jain KK. An assessment of zonisamide as an anti-epileptic drug. Expert Opin Pharmacother 2000 Sep; 1(6): 1245-60.
47) Kasteleijn-Nolst Trenité D.G.; Marescaux C.; Stodieck S.; Edelbroek P.M. Photosensitive epilepsy: a model to study the effects of antiepileptic drugs. Evaluation of the piracetam analogue, levetiracetam. Epilepsy res, 1996; 25(3): 225-230.
48) KERÄNEN T. & RIEKKINEN P. - Severe Epilepsy: Diagnostic and Epidemiological Aspects. Acta neurol. scand., 78(Suppl.117): 7-14, 1988.
49) KRAMER U.; NEVO Y.; NEUFELD M. Y.; FATAL A.; LEITNER Y.; HAREL S. - Epidemiology of epilepsy in childhood: a cohort of 440 consecutive patients. Pediatr Neurol 18: 46-50, 1998.
50) Leppik I.E., Gil-Nagel A. New antiepileptic drugs. In: Epilepsy. Ed. Pedley T.A., Meldrum B.S. Churchill Livingstone, 1995. p.119-139.
51) MILLER S. P.; DILENGE M. E.; MEAGHER-VILLEMURE K.; O’GORMAN A. M.; SHEVELL M. I. - Infantile epilepptic encephalopathy (Ohtahara syndrome) and migrational disorder. Pediatr Neurol, 19: 50-54, 1998.
52) Millichap J.G. Mechanisms of AED action. Pediatr neurol, 1991; 7(1): 72.
53) Minassian BA, Sainz J, Delgado-Escueta AV. Genetics of myoclonic and myoclonus epilepsies.Clin Neurosci 1995-96; 3(4): 223-35.
54) MOSHÉ S.L. - Seizures in the developing brain. Neurology, 43(Suppl. 5): S3- S7, 1993.
55) Nieto-Barrera M, Candau R, Nieto-Jimenez M, Correa A, del Portal LR. Topiramate in the treatment of severe myoclonic epilepsy in infancy.Seizure 2000 Dec; 9(8): 590-4.
56) Nieto-Barrera M, Lillo MM, Rodriguez-Collado C, Candau R, Correa A. Severe myoclonic epilepsy in childhood. Epidemiologic analytical study]. Rev Neurol 2000 Apr 1-15; 30(7): 620-4.
57) Nieto M, Roldan S, Sanchez B, Candau R, Rodriguez R. [Immunological study in patients with severe myoclonic epilepsy in childhood]. Rev Neurol 2000 Mar 1-15; 30(5): 412-4.
58) Oguni H, Fukuyama Y, Imaizumi Y, Uehara T. Video-EEG analysis of drop seizures in myoclonic astatic epilepsy of early childhood (Doose syndrome). Epilepsia 1992 Sep-Oct; 33(5): 805-13.
59) Oguni H.; Hayashi K.; Oguni M.; Mukahira A.; Uehara T.; Fukuyama Y.; Umezu R.; Izumi T.; Hara M. Treatment of severe myoclonic epilepsy in infants with bromide and its borderline variant. Epilepsia 1994; 35(6): 1140-1145.
60) Pelekanos J., Camfield P., Camfield C., Gordon K. - Allergic Rash Due to Antiepileptic Drugs. Clinical Features and Management. Epilepsia 1991; 32(4): 555-559.
61) PELLOCK J. M & APPLETON. - Use of new antiepileptic drugs in the treatment of childhood epilepsy. Epilepsia, 40(Suppl. 6): S29-S38, 1999.
62) Perez J, Chiron C, Musial C, Rey E, Blehaut H, d'Athis P, Vincent J, Dulac O. Stiripentol: efficacy and tolerability in children with epilepsy. Epilepsia 1999 Nov; 40(11):1618-26.
63) Pranzatelli M.R. & Nadi N.S. Mechanism of action of antiepileptic and antimyoclonic drugs. In: Advances in Neurology. Eds. Fahn S.; Hallett M.; Lüders H.O.; Marsden C.D. Lippincott-Raven. Cap. 23, 1996, p. 329-357.
64) Prasad A. N.; Stafstrom C. F.; Holmes G. L. Alternative epilepsy therapies: the ketogenic diet, immunoglobulins and steroids. Epilepsia 1996, 37(suppl.1), S81-S95.
65) REYNOLDS E.H. - The prevention of chronic epilepsy. Epilepsia, 29(Suppl.1): S25-S28, 1988.
66) REYNOLDS E.H.; ELWES R.D.C.; SHORVON S.D. Why does epilepsy become intractable. Lancet, 2: 952-954, 1983.
67) Reynolds E.H., Ring H.A., Farr I.N., Heller A.J., Elwes R.D.C. Open Double-Blind and LongTerm Study of Vigabatrin in Chronic Epilepsy. Epilepsia 1991; 32(4): 530-538.
68) SCHACHTER S.C. - Advances in the assessment of refractory epilepsy. Epilepsia, 34(Suppl. 5): S24-S30, 1993.
69) Shields W. D. Catastrophic epileps in childhhod. Epilepsia 2000; 41 (Sup[l. 2): S2-S6.
70) SILLANPÄÄ M. - Remission of seizures and predictors of intractability in long-term llow-up. Epilepsia, 34(5): 930-936, 1993.
71) SILVA E.A.; CHUGANI D.C.; MUZIK O.; CHUGANI H.T. - Identification of frontal lobe epileptic foci in children using positron emission tomography. Epilepsia, 38(11): 1198-1208, 1997.
72) SWINK T.; VINING E. P. G.; FREEMAN J. M. - The ketogenic diet. Adv Pediatr, 44: 297-329, 1997.
73) Tada H.; Morooka K.; Arimoto K.; Matsuo T. Clinical effects of allopurinol on intractable epilepsy. Epilepsia 1991; 32(2): 279-283.
74) Wallace SJ. Management issues in severe childhood epilepsies. Seizure 1995 Sep; 4(3):215-20.
75) WILLIAMS A. N.; GRAY R. G,; POULTON K.; RAMANI P.; WHITEHOUSE W. P. - A case of Ohtahara syndrome with cytocrome oxidase deficiency. Dev Med Child Neurol ., 40: 568-570, 1998.
76) WYLLIE E.; CHEE M.; GRANSTRÖM M.L.; DEL GIUDICE E.; ESTES M.; COMAIR Y.; PIZZI M.; KOTAGAL P.; BOURGEOIS B.; LÜDERS H. - Temporal lobe epilepsy in early childhood. Epilepsia, 34(5): 859-868, 1993.
77) Woody R.C. Bromide therapy for pediatric seizure disorder intractable to other antiepileptic drugs. J child neurol 1990; 5 (1): 65-67.
78) Zagnoni P.G.; Bianchi A.; Zolo P.; Canger R.; Cornaggia C.; D’Alessandro P.; De Marco P.; Pisani F.; Gianelli M.; Verzé L.; Viani F.; Zaccara G. Allopurinol as add-on therapy in refractory epilepsy: a double-blind placebo-controlled randomized study. Epilepsia 1994, 35(1): 107-112.

Destaques
Notícias
mais notícias...